|
NuFold Database
|

|
A large-scale structural resource of over 545,000 RNA tertiary structures predicted by our NuFold. It addresses a critical gap in RNA biology by offering structural coverage for over 2,200 RNA families, featuring per-nucleotide pLDDT and a novel secondary structure agreement score (secF1) to ensure model reliability.
|
|
|
PFP Server
|

|
Unified protein function prediction server that combines PFP, ESG, PhyloPFP, and DomainPFP.
|
|
|
Queryome
|

|
Queryome is an agentic deep research assistant that helps researchers explore biological literature. It can answer questions, summarize papers, and find relevant information from the vast amount of biological literature. Visit the web server to try it out.
|
|
|
NuFold
|

|
NuFold is a state-of-the-art method designed for predicting 3D RNA structures, leveraging deep learning for high accuracy and reliability.
|
|
|
EM Server
|

|
Web server of our popular tools for cryo-EM structure modeling and validation, including DeepMainmast, CryoREAD, DAQ, and DAQ-Refine.
|
|
|
EMSuites
|

|
The suite of programs developed by our group for accurately identifying protein secondary structures. (Emap2Sec), protein nucleic acid (RNA/DNA) (Emap2Sec+), full atom 3D model MAINMAST or segmentation of individual components (MainMastSeg), improved protein modeling from GAN-modified EM maps (EM-GAN), model quality assessment with (DAQ Score), model quality assessment results in (DAQ-Score Database), local model refinement with (DAQ-refine), real-time comparison and analysis of cryo-Electron Microscopy (EM) maps (EM-Surfer), and EM map superimposition (VESPER).
|
|
|
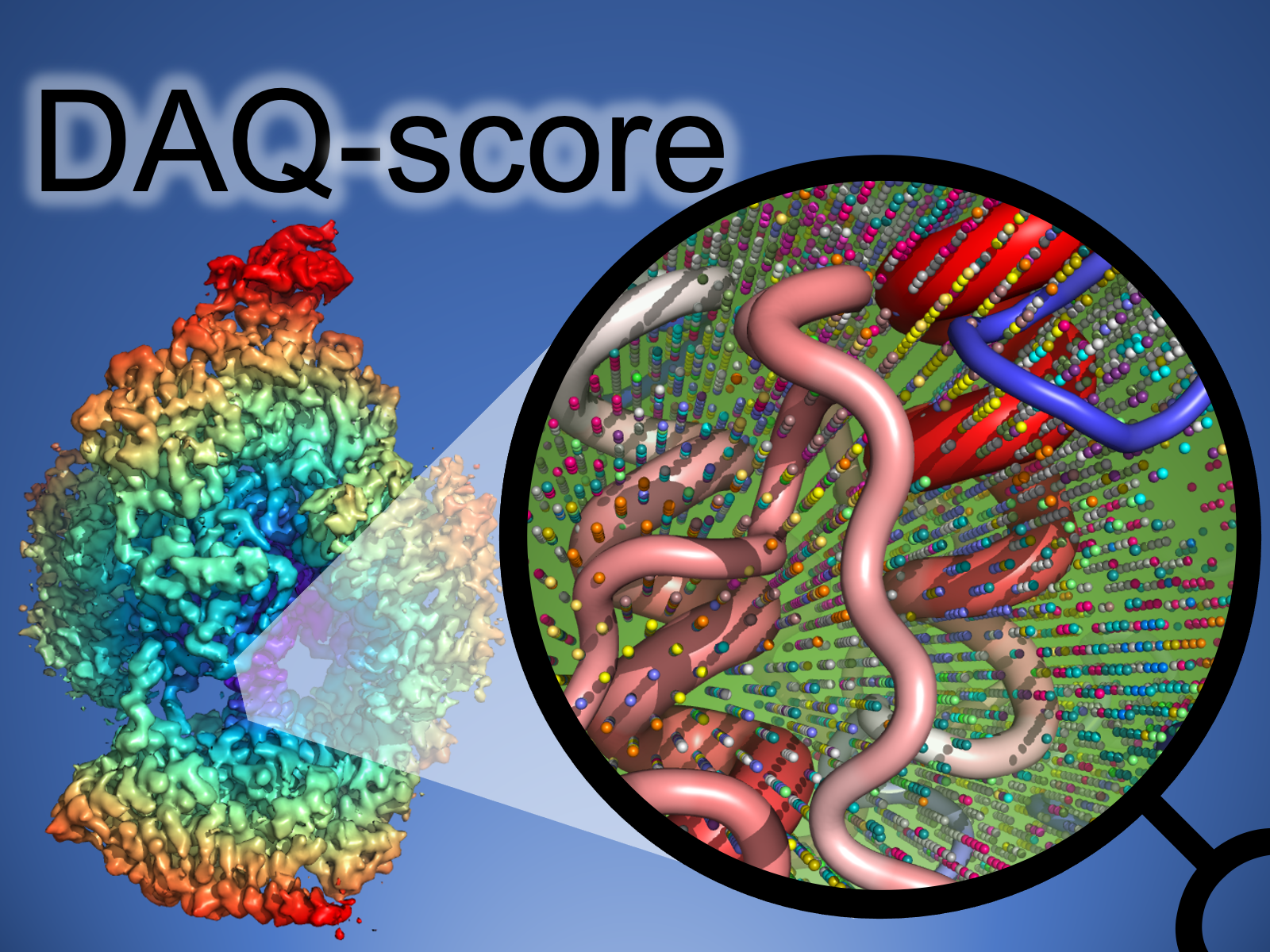
DAQ
|
|
DAQ Score is a computational tool using deep learning that can estimate the residue-wise local quality for protein models from cryo-Electron Microscopy (EM) maps.
|
|
|
DAQ-Score Database
|

|
DAQ-Score Database stores precomputed residue-wise local quality scores of protein models from cryo-EM maps.
|
|
|
DAQ Refine
|
|
DAQ Refine is a new protocol for evaluating a protein model built from a cryo-EM map and applying local structure refinement in the case where the model has potential errors.
|
|
|
Emap2Sec
|
|
Emap2sec is a deep learning-based tool for detecting protein secondary structures from intermediate resolution cryo-EM maps. Visit the website, the Github site, or the paper on Nature Methods.
|
|
|
MainMast
|

|
MAINMAST is a de novo modeling protocol to build an entire protein 3D model directly from near-atomic resolution EM map. Visit the web site or read the paper on Nature Communications
|
|
|
MAINMASTseg
|

|
MAINMASTseg is a minimum spanning tree based segmentation program for EM density map. Visit the web site.
|
|
|
LZerD Web Server
|

|
The LZerD web server provides easy access to our LZerD protein-protein docking software suite through an intuitive web interface. Visit the website.
|
|
|
LZerD Software Suite
|

|
The protein docking suite developed includes programs to perform protein-protein docking prediction, multiple protein docking, as well as protein docking prediction using predicted protein-protein interfaces. The Linux binaries can be downloaded here.
|
|
|
Dove
|

|
Dove is a deep learning based protein docking model evaluation method. It will use the atom information such as positions, types, energy scores in the interface area to judge if the docking model is reasonable. You can use the server here or download source code to run by yourself.
|
|
|
BindML
|

|
BindML predicts protein-protein interaction sites in a query protein structure by using evolutionary information. BindML+ further classifies a binding site to permanent and transient interaction.
|
|
|
3DSurfer 2.0
|

|
3D-SURFER 2.0 is a web platform for real-time protein surface comparison of a given protein structure against the entire PDB using 3D Zernike descriptors. It can compare the protein surface of a single chain, domain, or complex against databases of protein chains, domains, complexes, or a combination of all three, in the latest PDB. The databases are weekly updated. Visit the server or read the paper.
|
|
|
EM-Surfer
|

|
EM-SURFER is a web platform for real-time electron microscopy database search. It compares isosurface shape of a query EM map against maps in the latest EMDB. The databases are weekly updated. Visit the server or read the paper.
|
|
|
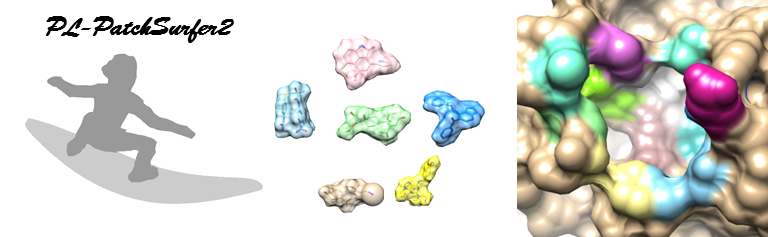
PL-PatchSurfer2
|
|
PL-PatchSurfer2 is a protein-ligand virtual screening program. The program searches a binding candidate ligand of a target protein by comparing local surface patches. The binary file can be downloaded here.
|
|
|
PatchSurfer 2.0
|
|
PatchSurfer2.0 predicts binding ligand(s) for a query protein pocket by comparing local surface patches of known ligand binding pockets to the query. This is the trial version associated with the publication.
|
|
|
PhyloPFP
|

|
PhyloPFP is a phylogenomics based protein function prediction server. Visit the server to submit a sequence.
|
|
|
ESG
|

|
ESG is a sequence similarity-based protein function prediction server. It employs PSI-BLAST iteratively and essentially selects GO term annotations that appear consistently in the searches. Visit the server to submit a sequence or read the paper. ESG was among top in the 1st CAFA function prediction assessment.
|
|
|
PFP
|

|
PFP is a sequence similarity-based protein function prediction server designed to predict GO annotations for a query sequence beyond what can be found by conventional database search such as BLAST. It takes into account weakly similar sequences as well as GO term associations observed in known annotations. Visit the server to submit a sequence or read the paper. PFP had the highest total score in a function prediction contest held at AFP-SIG'05 (ISMB2005), and also was the best in the function prediction category at CASP7.
|
|
|
PFP + ESG
|

|
Combined PFP+ESG interface
|
|
|
NaviGO
|
|
Interactive Gene Ontology visualization and similarity computation.
|
|
|
COVID19 Structure Modeling
|

|
Protein structure modeling for SARS-CoV-2.
|
|
|
GMQ
|
|
GMQ is a protein quality assessment program which employs conditional random field. The program gives a binary prediction that predicts a modeled residue has an error within a C-alpha distance cutoff or not.
|
|
|
FlexPred
|

|
FlexPred predicts real-value fluctuation of each residues in a query protein structure. Predictions can be performed for single-chain, complex proteins, and computational models.
|
|
|
DextMP
|
|
DextMP Text-mining tool to find Moonlighting Proteins from publication titles, abstract and functional description in UniProt.
|
|
|
MPFit
|

|
MPFit Predicts Moonlighting Proteins from Gene Ontology (GO) and diverse omics-based protein association features.
|
|
|
VisGrid
|

|
VisGrid identifies pockets in protein surfaces. The algorithm computes open directions, named visibility, at surface points. The paper is available at PubMed and also from the publication list.
|
|
|
GOMEP
|

|
GOMEP Predicts Missing Enzymes using functional coherence scores of Gene Ontology (GO) terms.
|
|
|
MASQ PPI
|
|
The purpose of MAss spectrometry data and Protein SeQuence-based PPI prediction method (MASQ_PPI) is to develop a complementary computational approach to aid large-scale protein-protein interactions (PPIs) identification using mass spectrometry data. The protein elution profiles are generated from size-exclusive chromatography followed by mass spectrometry. Proteins are clustered based on their elution profile. Then sequence-based computational prediction method are performed on the protein pairs from the same cluster by support vector machine to exclude some false positive interactions. Visit the code on Github.
|
|
|
SUPRB threading program(v1.0)
|
|
SUPRB is a threading structure prediction algorithm which employs suboptimal alignments. Supplementary material for paper - Effect of Using Suboptimal Alignments in Template-Based Protein Structure Prediction. Hao Chen and Daisuke Kihara. Proteins, 2010.
|
|
|
Suboptimal Alignment(v1.0)
|
|
Suboptimal Alignment is the protein structure prediction program based on threading strategy with SPAD, the error estimator, provided as supplementary material for paper - Estimating Quality of Template-Based Protein Models by Alignment Stability. Hao Chen and Daisuke Kihara. Proteins, 2008. Visit the server
|
|
|
EMD 1.0 (Ensemble Motif Discovery)
|
|
EMD (Ensemble Motif Discovery) is an ensemble (consensus) algorithm that identifies one or more frequent motifs among multiple sequences. The basic idea is to combine motif predictions from multiple runs of multiple component algorithms to build consensus motifs as its prediction. In the current version, five component algorithms are included - AlignACE, BioProspector, MotifSampler, MDScan, and MEME. The former three are stochastic algorithms, while the latter two are deterministic algorithms. Visit the server
|
|