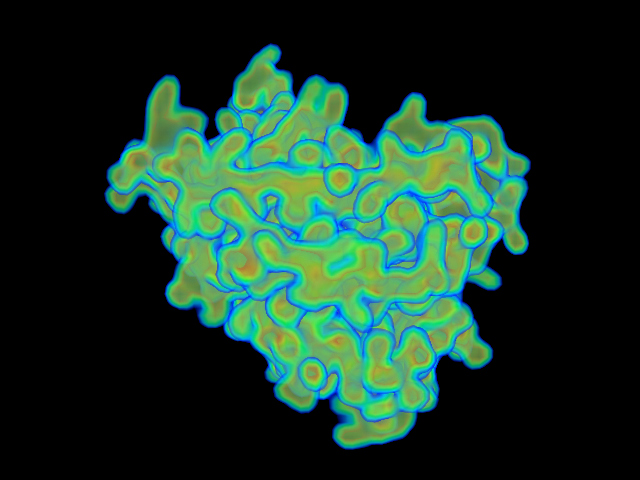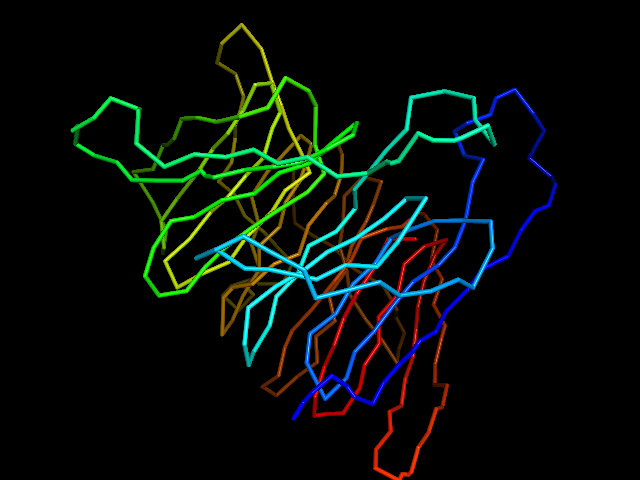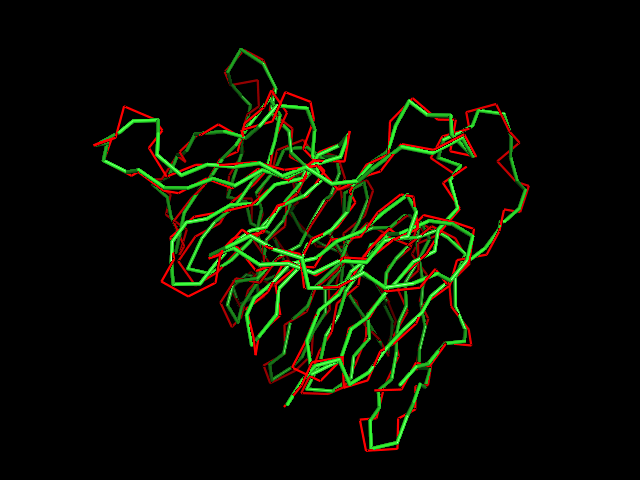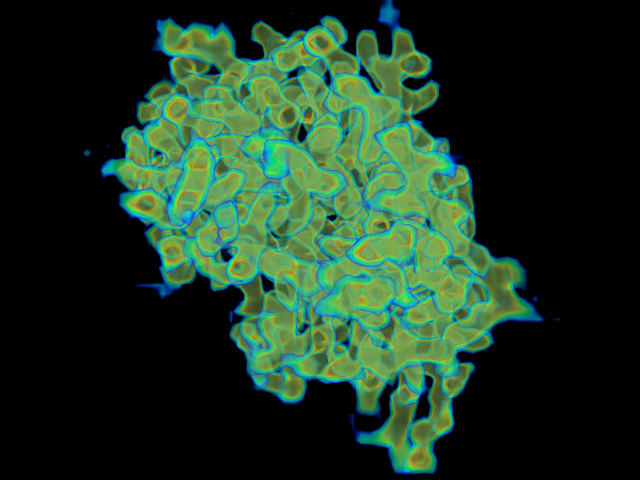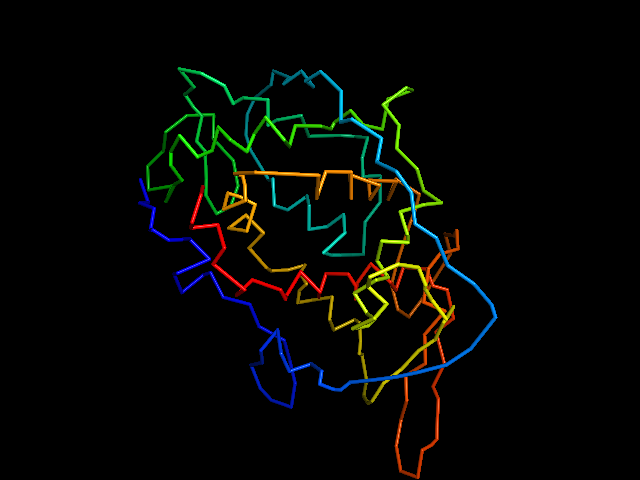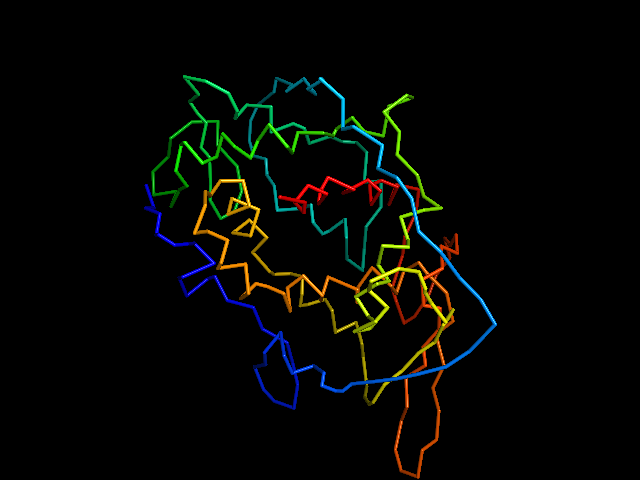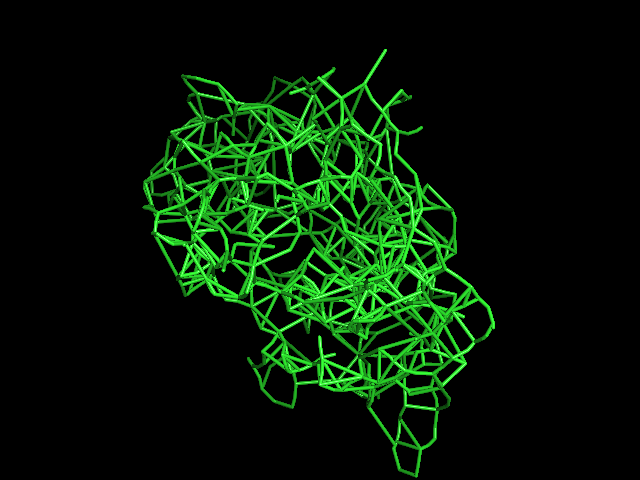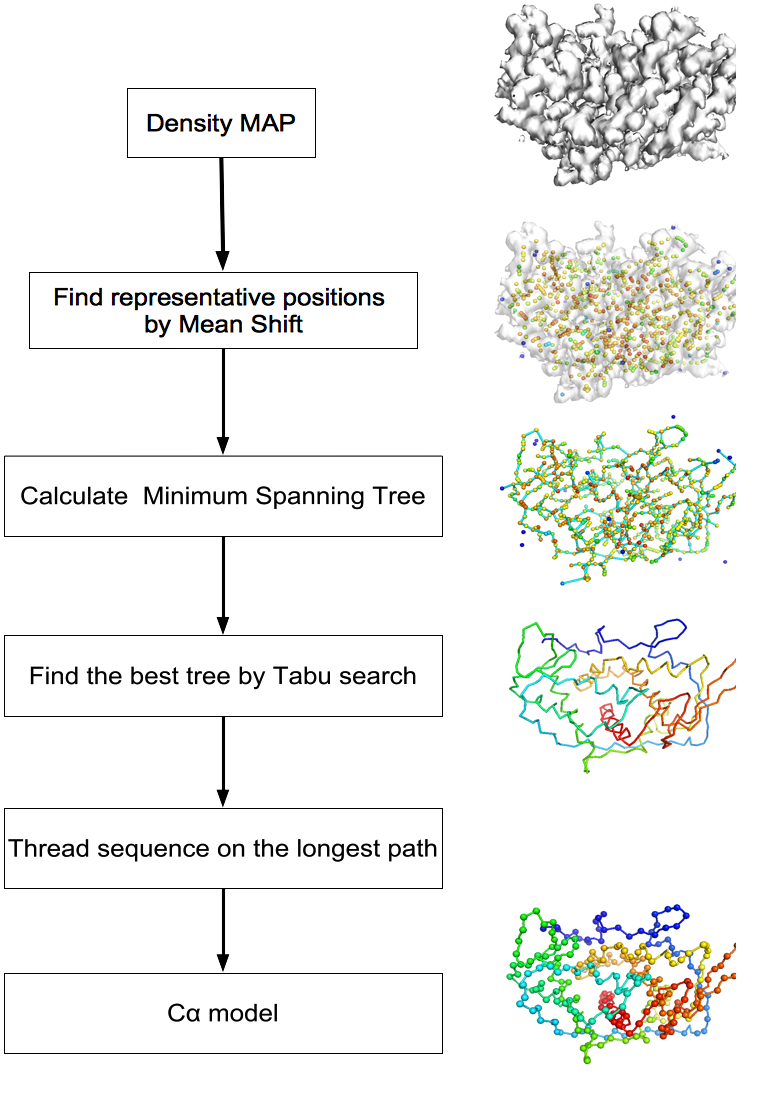
 MAINMAST protocol
MAINMAST protocol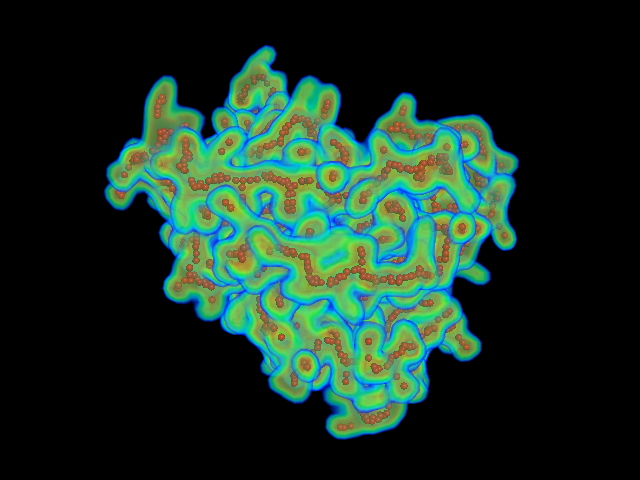
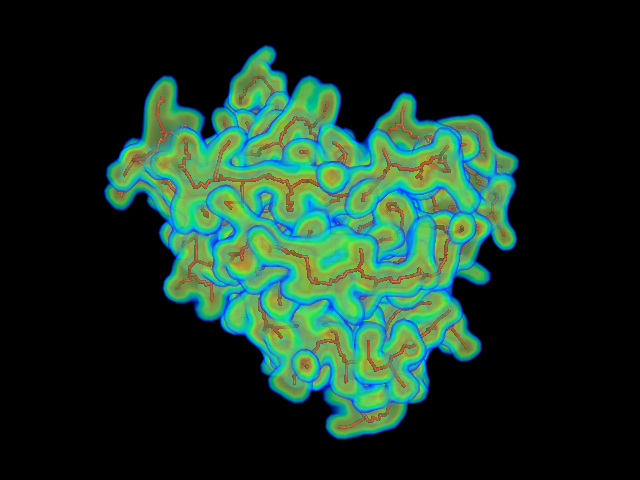
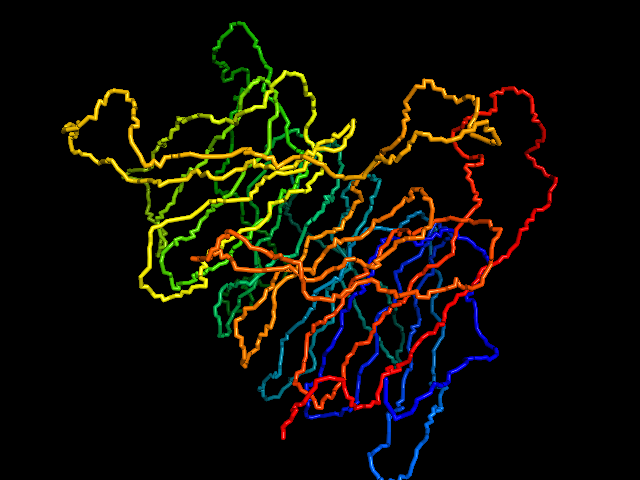
MAINMAST
This command identify local dense points (LDPs) in an EM map. Then LDPs are connected by Minimum Spanning Tree (MST). The MST is refined by a tabu search algorithm.
Usage: MAINMAST -m [MAP file (situs format)] (option) Option ver2.0: -Tree : Show MSTree mode -Graph : Graph mode ---Parameters in MeanShift---- -gw [f] : bandwidth of the gaussian filter def=2.0, sigma = 0.5*[float] -Dkeep [f] : Keep edge where distance < [f] def=0.5 -t [f] : Threshold of density values. def=0.0 -allow [f] : Max shift distance < [f] def=10.0 -filter [f]: Filter of representative points def=0.1 -merge [f]: After MeanShifting, merge d<[f] def=0.5 ---Parameters in Tabu-search---- -Nround [i]: Number of Iterations def=5000 -Nnb [i]: Number of Neighborss def=30 -Ntb [i]: Size of tabu-list def=100 -Rlocal [f]: Radius of Local MST def=10 -Const [f]: Constraint of total length of edge def=1.01,Total(Tree) <[f]*Total(MST)