
DEEPMAINMAST
DEEPMAINMAST is a de novo modeling protocol to build
an entire protein 3D model directly from near-atomic resolution EM map.
DEEPMAINMAST employs deep learning to
capture the local map features of amino acids and atoms to assist main-chain tracing. We also integrated the protocol with Alphafold2 to achieve even higher accuracy.
Additionally, the protocol is able to accurately assign chain identity to the structure models of homo-multimers.
Tutorial Video from DAQ & DeepMainmast Workshop is made available at our lab channel.
Introduction
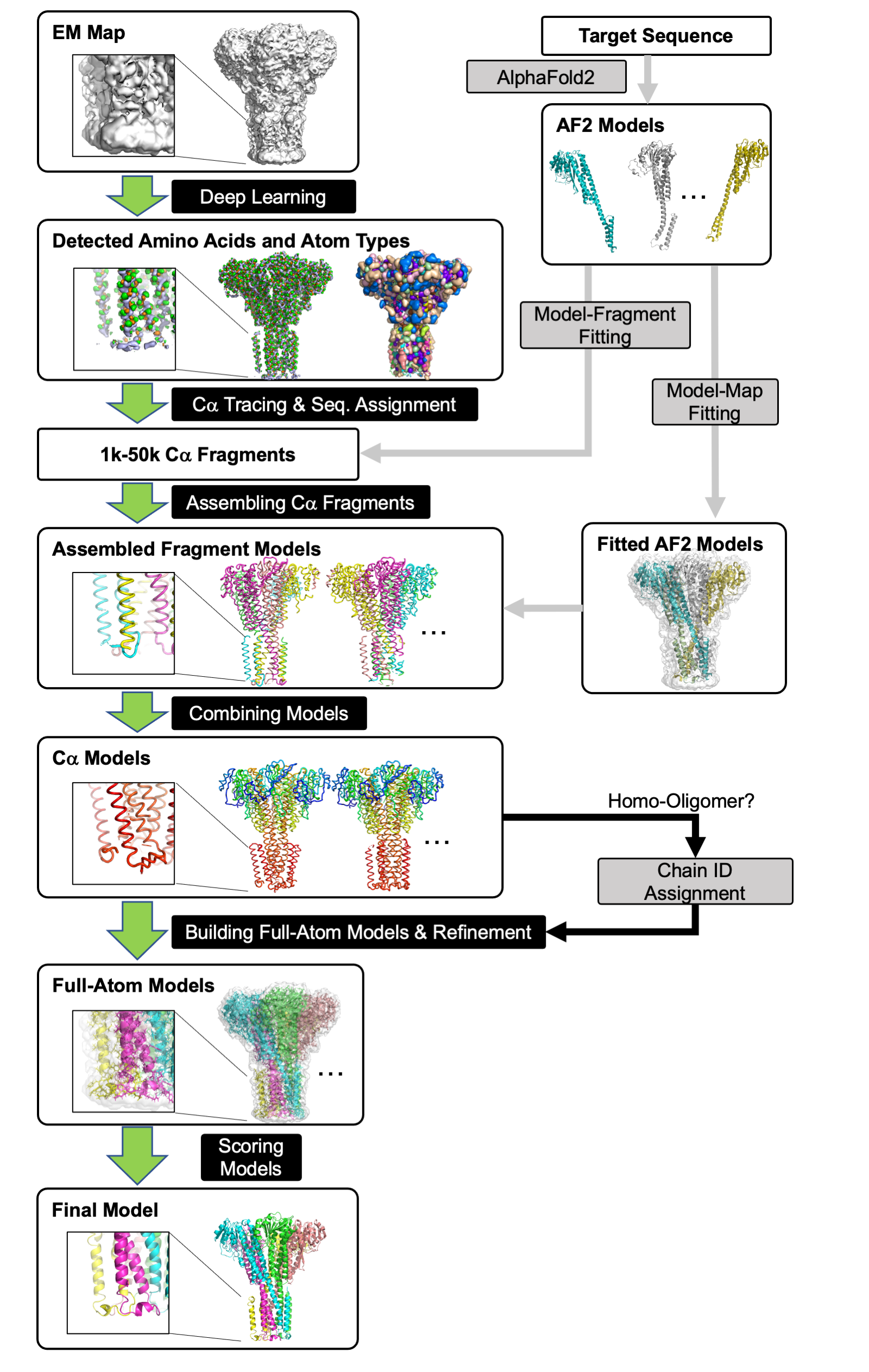
The DeepMainmast Protocol
The green arrows represent the core of the protocol, DeepMainmast(base). It consists of six logical steps:
- 1) Detecting amino-acid types and atom types using deep learning (Emap2sf). The image on the left shows the detected atom types (Cα atom: green, carbon: orange, and nitrogen: light blue). The image on the right shows the detected amino acid types in different colors.
- 2) Tracing Cα path and assigning the target sequence using the Vehicle Routing Problem Solver and the Dynamic Programming algorithm. Different parameter combinations are used.
- 3) Assembling Cα fragments using the Constraint Problem (CP) Solver. Colors indicate chain IDs.
- 4) Combining Cα models built under different parameter combinations using the CP Solver. Colors indicate the direction of chains from blue to red for the N-terminal to the C-terminal residues.
- 5) Full-atom building and refinement using PULCHRA and Rosetta-CM.
- 6) Scoring generated full-atom models based on the DAQ(AA) score and the DOT score. For homo-oligomer targets, chain IDs are assigned based on the structural similarity of homomer proteins (black arrows).
- 7) The gray arrows represent the procedure to use AF2 models. AF2 models were integrated into the modeling process in two ways, (i) Imposing fragments in the AF2 models to Cα fragments; and by (ii) Fitting AF2 models to the EM map using Map-Model fitting program, VESPER.
Usage guide
Free Server (Recommended): https://em.kiharalab.org/algorithm/DeepMainMast
Tech Specs
CPU: >=4 cores
Memory: >=64Gb (for small structures with less than 1000 residues, 32GB should be enough)
GPU: any GPU supports CUDA with more than 12GB memory.
Run on your local computer
First go to the github repo and then clone it to download on your local machine. Follow the readme instruction or the the following instructions`
There are two folders i) dmmsinglechain supportd single chain protein maps ii) dmmmultichain supports multi chain protein maps.
1. Dependencies
Use the requirements.txt file and install packages using pip as follows. There are same requirements for both single chain and multi-chain.
pip install -r requirements.txt
Then install BioTEMPy package using pip. Installing it separately is crucial because it has dependecy conflicts with bio package. Hence after installing all packages
using requirements.txt, we then separately install BioTEMPy using pip as follows
pip install BioTEMPy
2. Execute the DeepMainMast command
Next, follow the details of the instructions by reading the readme file.
dmm_full_multithreads.sh: DeepMainmast Multithreads with Full-Atom refinement by RosettaCM and AF2 model.
Other easier ways to run quickly
Google Colab Notebooks:
Code Ocean Capsule:
Server:
Running our code ocean capsules
1. First, you need to create an account on code ocean using academic credentials and login into your account.
2. Then, click on the links above and go to the desired code ocean capsule.
3. To make a reproducible run i.e. run the code on an example input provided by us, click on the "Reproducible Run" button in the top right corner. This will start running the code on our example, and the results will be generated in the results folder at the bottom right after the execution is complete.
4. To run the code on an input of your choice, first go to our capsule and click on the "Edit Capsule" button in the top right corner. This will make a copy of our capsule which you can edit. Follow the instructions about how to upload and run your input in this copied capsule by reading the readme file present in the respective code ocean capsules.
Other details specific to the respective capsules can be found in the readme files of the capsules. For more details on how to run code ocean capsule please visit: Code Ocean user documentation
Examples of output files
We provide some sample output files prodcued by DeepMainMast when we ran it on our examples. examples
License
© 2023 Genki Terashi, Xiao Wang, Devashish Prasad, Daisuke Kihara and Purdue University
DEEPMAINMAST is a free software for academic and non-commercial users.
It is released under the terms of the GNU General Public License Ver.3
(https://www.gnu.org/licenses/gpl-3.0.en.html).
Commercial users please contact dkihara@purdue.edu for alternate licensing.
Reference
Citation of the following reference should be included in any publication that uses data or results generated by DEEPMAINMAST program.
(a)
Genki Terashi, Xiao Wang, Devashish Prasad, Tsukasa Nakamura & Daisuke Kihara. DeepMainmast: Integrated protocol of protein structure modeling for cryo-EM with deep learning and structure prediction. Nature Methods, 21: 122-131 (2024)
(b)
Integrated Protocol of Protein Structure Modeling for Cryo-EM with Deep Learning and Structure Prediction,
Genki Terashi, Xiao Wang, Devashish Prasad, Tsukasa Nakamura, and Daisuke Kihara, BiorXiv (2023)
© 2026 KIHARA Bioinformatics LABORATORY, PURDUE University | Design by TEMPLATED.