
| Home | Server | LZerD | Multi-LZerD | PI-LZerD | IDP-LZerD | Path-LZerD | Flex-LZerD | About |
|
|
|||||||||||||||||||
|
|
Multi-LZerD: Multiple protein docking for asymmetric complexes
Updates
Introduction Until now most of the protein docking prediction methods have focused on pairwise docking. There exist a handful of methods for the prediction of multimeric complexes, but almost all of them assume specific properties such as homomericity or symmetry. Considering a substantial number of multimeric complexes of diverse kinds exist in a cell, there is an urgent need for the development of a multiple protein docking prediction method that does not target a specific type of complexes. We have developed a novel computational multiple protein docking algorithm, Multi-LZerD, that builds models of multimeric complexes by effectively reusing pairwise docking predictions of component proteins. Overview of Multi-LZerD Multi-LZerD can be applied when users need to generate models of more than two proteins. In the first phase, pairwise docking predictions are generated for every possible pair of protein units, using LZerD. Then, a set of randomly generated graphs are created to represent different ways in which the units can bind to each other, in order to create the larger multimeric complex. For each pairwise connection created, a docking pose between the two proteins involved is selected randomly from LZerD predictions from the first phase. The graphs are then subject to an iterative improvement via a Genetic Algorithm (GA) that uses a physics-based fitness function. Finally, after a configurable number of iterations, a final refinement step is applied to the structures. Multi-LZerD was able to handle different graph topologies, as well as outperform the only method that could be directly compared to it, particularly in unbound docking cases. Download Multi-LZerD The current version of Multi-LZerD is available as a tar.gz file and can be downloaded here. Execution instructions are given in the README file provided. The dataset used for evaluation can be downloaded here. Both bound and unbound cases are included and a detailed description is provided in the README file. Physics based scoring function The physics-based scoring function is provided as a standalone program here. The README file explains how to generate the score for a PDB file that contains two or more interacting units.Executing Multi-LZerD with multiple threads
Executing on a single thread, Multi-LZerD may take a prohibitively long time to execute. The Multi-LZerD executable will automatically try to take advantage of multithreading if your system supports it. To manually control the number of threads Multi-LZerD attempts to use, set the
General Information Download and save the tar files of the program in your preferred directory. The files can be unpacked using the commands:
The program hbplus is compiled on x86-32bit. If you use a Windows Subsystem for Linux, the source code may be available at https://github.com/mmravic314/bin/blob/master/HBplus . The program has as default “basename: ‘sample’ ” and “units: ‘A, B, C’ ”. Make sure to modify “run.sh” and change these according to the files you are using. For example, for the protein 1A0R we would have:
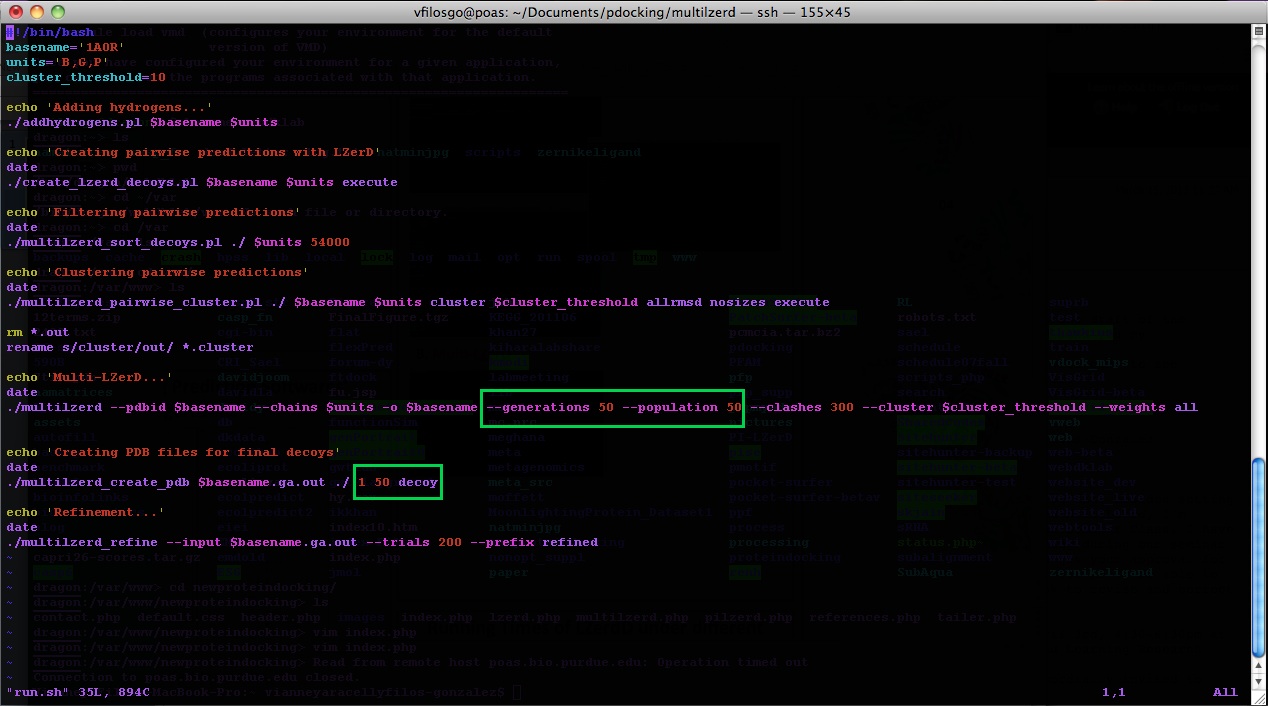
Program Output
When the program starts running, it will first read in the PDBs and write “.pdb.h” files, Ex:
|
| Copyright © 2026 KIHARA Bioinformatics LABORATORY, PURDUE University | |