| Home | Server | LZerD | Multi-LZerD | PI-LZerD | IDP-LZerD | Path-LZerD | Flex-LZerD | About |
|
|
|||||||||||||||||||
|
|
LZerD: Protein-Protein Docking Algorithm Updates
Introduction Existing docking methodologies generate anywhere from a few hundred to thousands of candidate complexes, each with a score that helps the users determine what models are considered to be better. Most of the existing methodologies deal with pairwise docking, where one protein binds to another one. LZerD (Local 3D Zerike descriptor-based Docking algorithm) is a program that uses geometric hashing to generate ligand orientations using the 3D Zernike descriptor as the shape matching criteria. Overview of LZerD LZerD, our pairwise docking program, can create protein complex structures from two individual structures (called receptor and ligand). It uses a combination of geometric hashing and a rotation invariant mathematical surface shape representation, the 3D Zernike Descriptors (3DZD), as the main tools to create putative docking poses. LZerD showed that the 3DZD representation is suitable to assess protein-protein interface similarity and generated good results when tested against docking benchmark cases and compared favorably against existing protein-protein methods. Download LZerD The current version of LZerD as used in the paper is available as a tar.gz file and can be downloaded here.Tutorial Getting Started Download and save the tar file of the program in your preferred directory. The files can be unpacked using the commands:
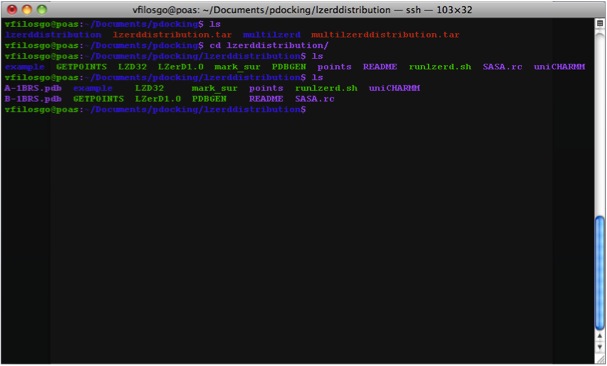
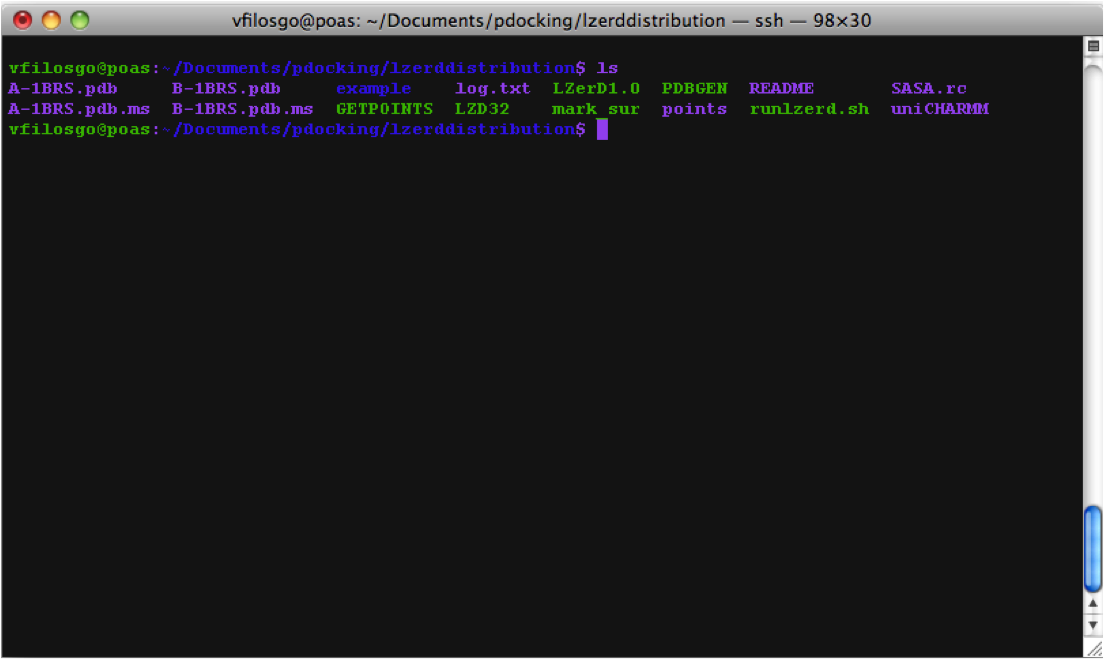
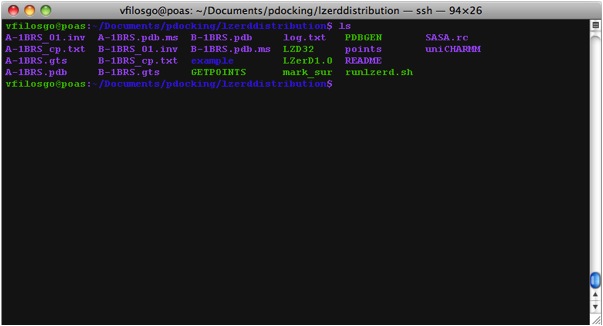
What The Directory Looks Like The generated Directory contains the files:
Copy your PDBs to the right directory; that is, the directory named “lzerddistribution”.
Running the Program To run the program you use the runlzerd.sh script providing it with two PDB files.
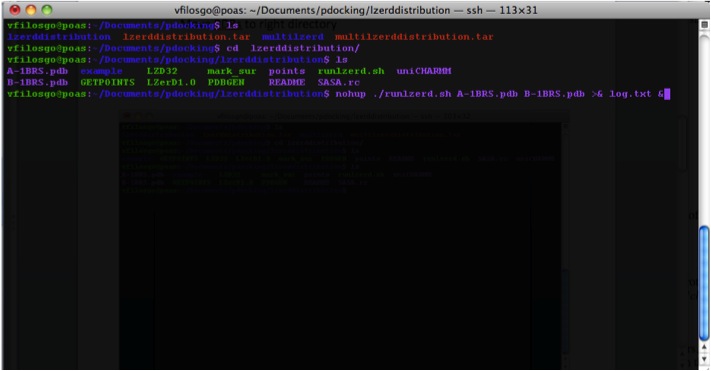 First the program generates .ms files (at this point, no output is visible in the terminal):
 Calculating Surfaces The program's first terminal output starts with the Calculation of surfaces: 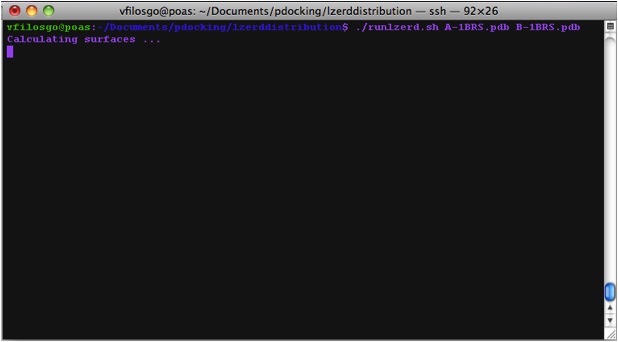
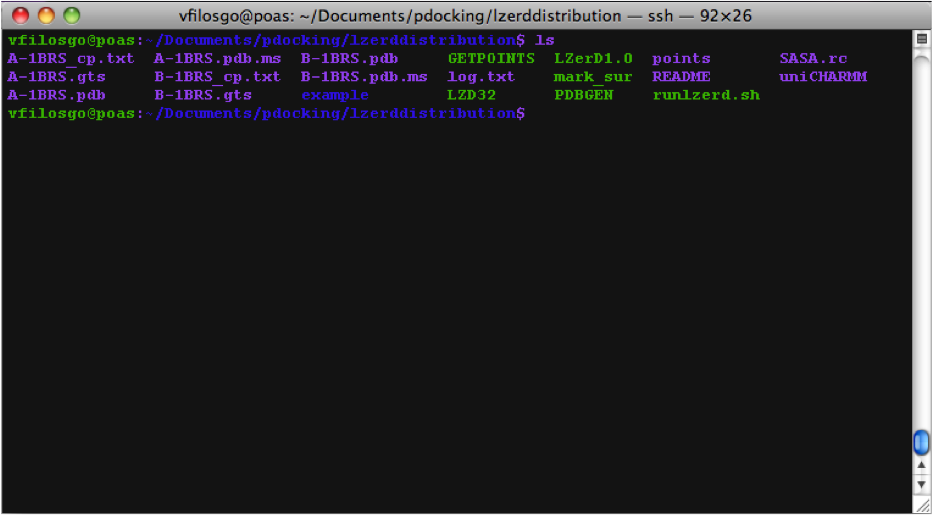 Calculating Zernike Descriptors 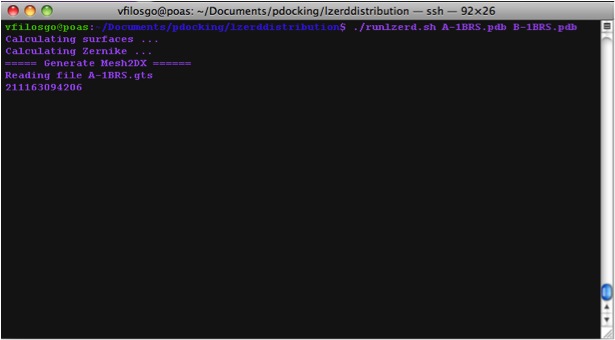 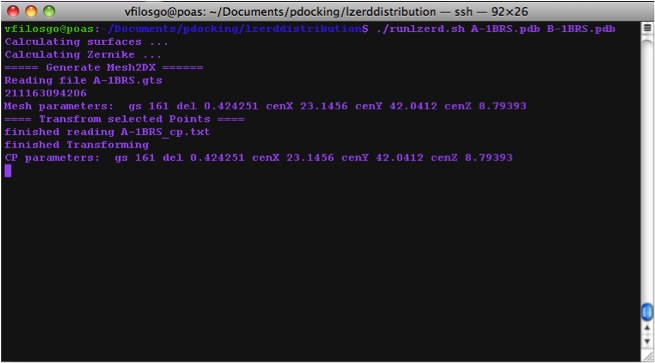 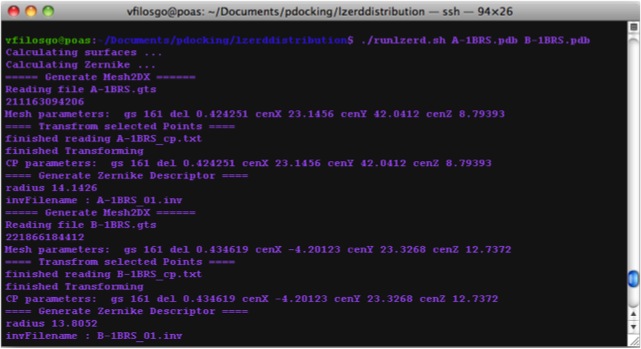
 Now we are Running LZerD 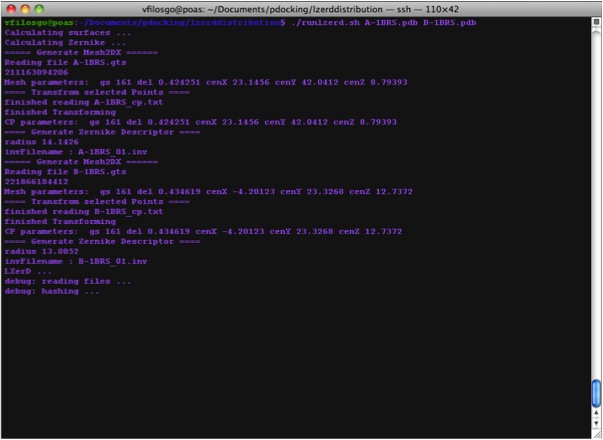
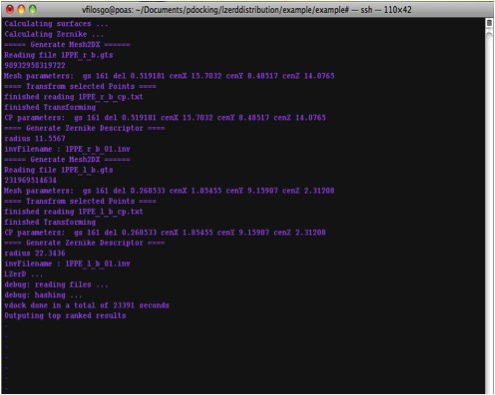
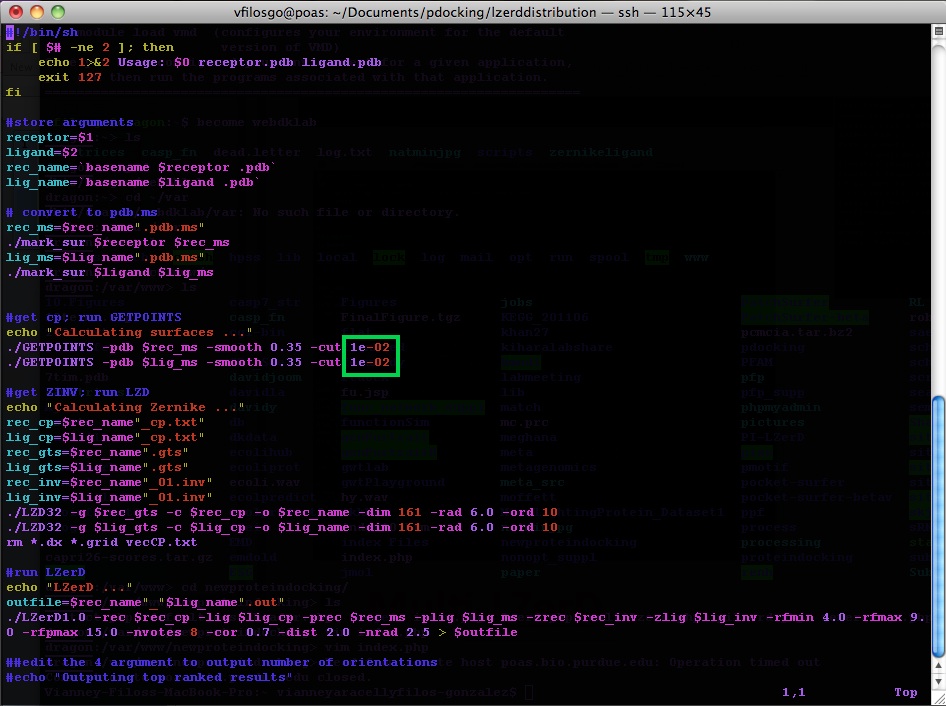 At the termination of the program, it would output the time it took to finish
 |
| Copyright © 2026 KIHARA Bioinformatics LABORATORY, PURDUE University | |