
| Home | Server | LZerD | Multi-LZerD | PI-LZerD | IDP-LZerD | Path-LZerD | Flex-LZerD | About |
|
|
|||||||||||||||||||
|
|
IDP-LZerD: Intrinsically Disordered Protein Docking Download IDP-LZerD from GitHub.Introduction Disordered protein-protein interactions (disordered PPIs), which involve an intrinsically disordered protein (IDP) are typically considered outside the scope of conventional docking. Protein-peptide docking methods exist, but they are generally designed for very short protein chains. IDP-LZerD, our disordered PPI modeling method, follows the dock-and-coalesce model of disordered PPI formation. The problem is brought within the scope of conventional docking by initially docking IDP fragments to the receptor, followed by a combinatorial path search and model refinement. IDP-LZerD has been demonstrated to work on IDPs up to 69 residues long. Inputs IDP-LZerD requires the sequence of the IDP to be modeled and a PDB-format model of the receptor structure. The receptor model can be either an unbound or bound structure. IDP-LZerD also accepts secondary structure prediction methods, and it is recommended to input any confident secondary structure information available. Example of secondary structure prediction methods include:
Outputs IDP-LZerD outputs full-atom models of the disordered PPI. Methods
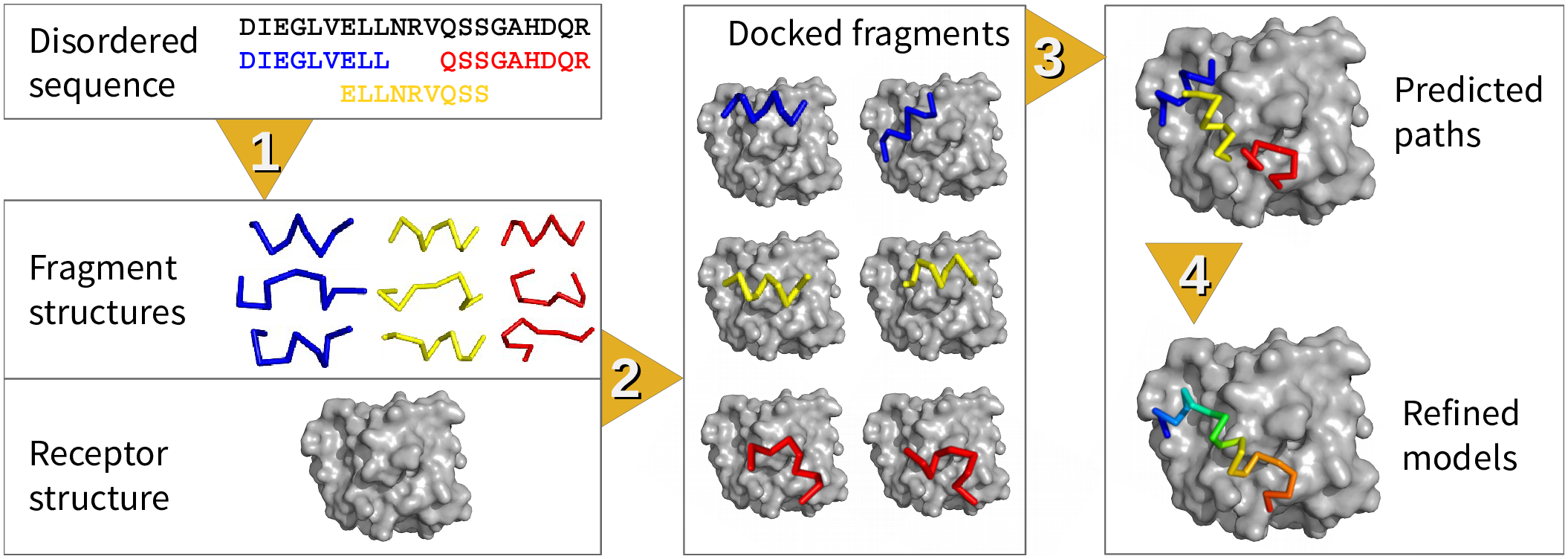
1. fragment structure prediction, 2. fragment docking, 3. path assembly, and 4. refinement. Steps 1 and 2 correspond to “dock” and Steps 3 and 4 correspond to “coalesce.” This figure is taken from Figure 1 of the original IDP-LZerD paper [1] (available under the Creative Commons CC0 public domain dedication). 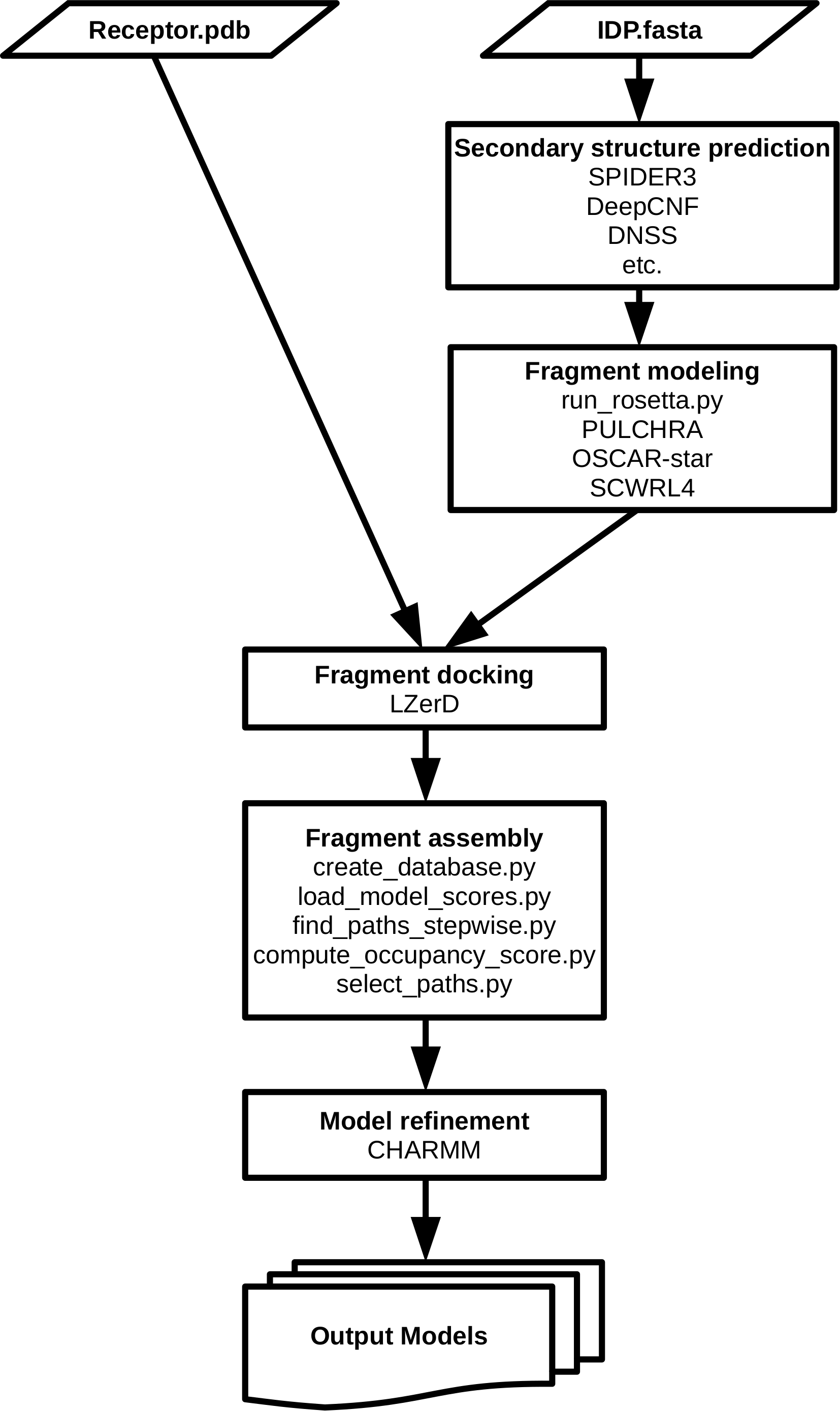
The above figures show the general flow of IDP-LZerD. Given a receptor structure and an IDP sequence, full-atom fragment models of the IDP are generated using the Rosetta fragment picker, possibly including secondary structure information from secondary structure prediction methods or from the literature. Then, our protein-protein docking software, LZerD, is used to dock the fragment models to the receptor. The docked fragments are then combinatorially assembled into full-chain models, and are then finally refined using a short molecular dynamics simulation. References
If you use IDP-LZerD, please cite:
|
| Copyright © 2026 KIHARA Bioinformatics LABORATORY, PURDUE University | |