Kihara Bioinformatics Laboratory
Our research interests lie in the area of bioinformatics. We employ computational methods to elucidate intertwined relationships between protein/gene sequences, structure, function, interactions, genome, and pathways. The ultimate goal of our research projects is to obtain new, comprehensive understanding of how structures and functions are coded in molecular sequences and how functions of molecules are orchestrated in a cell.
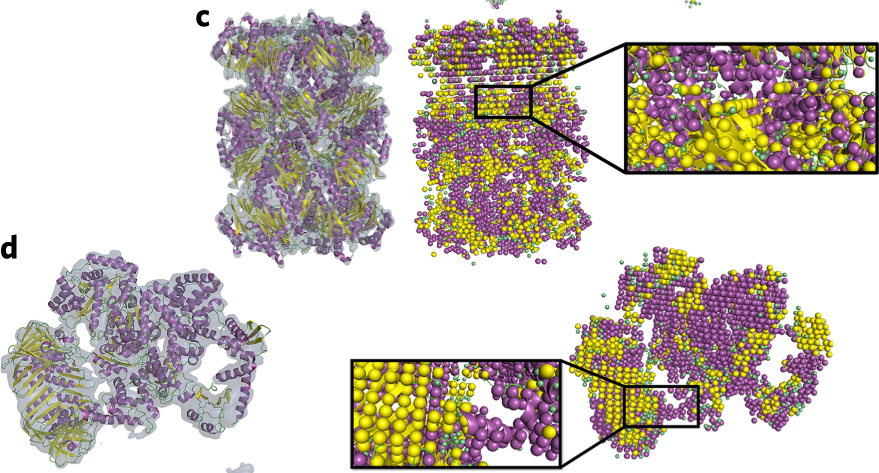
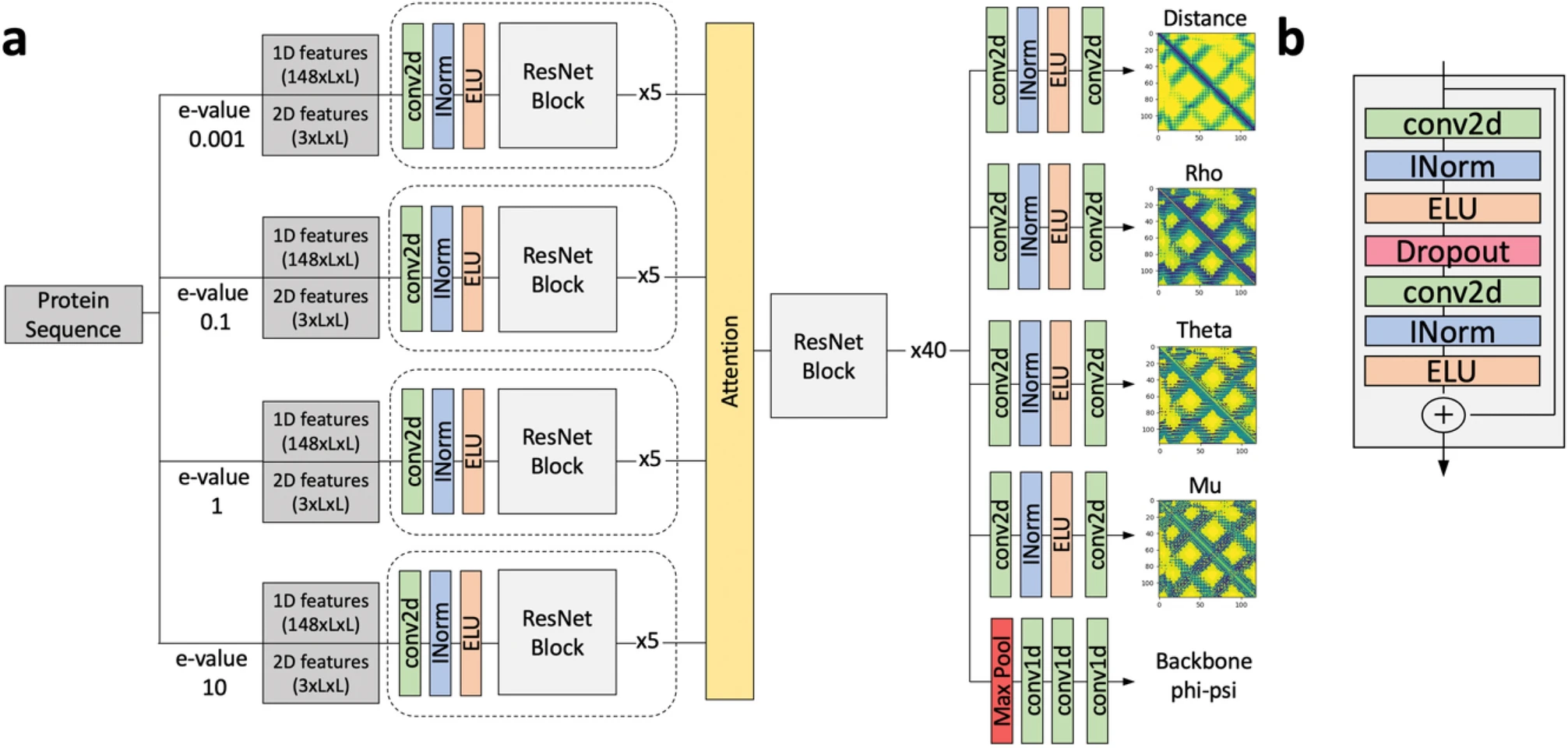
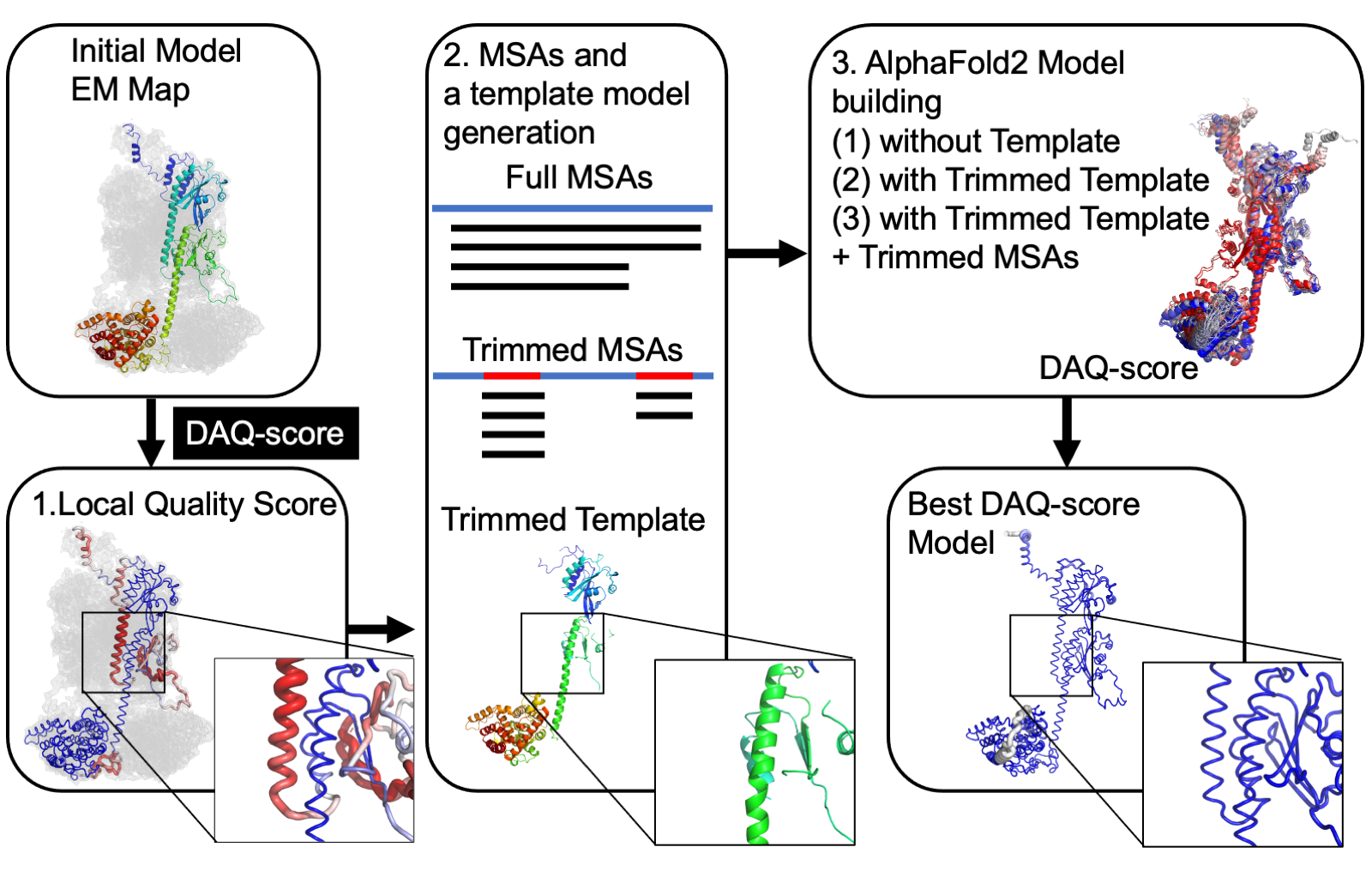
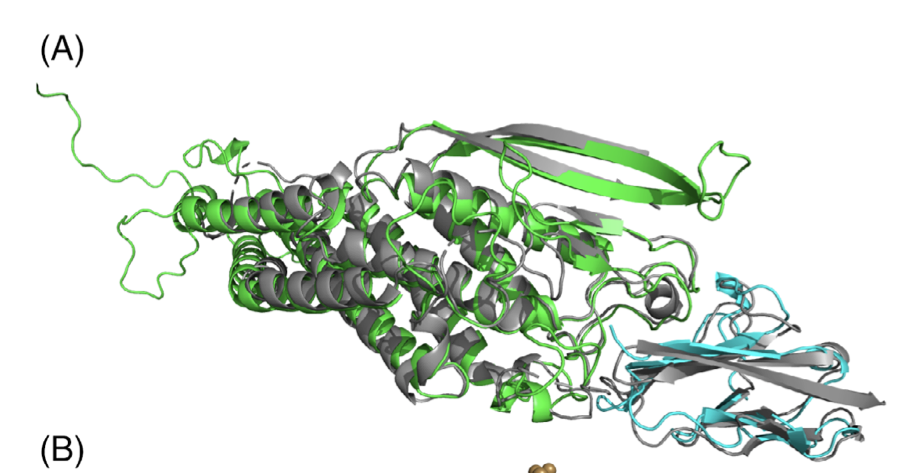
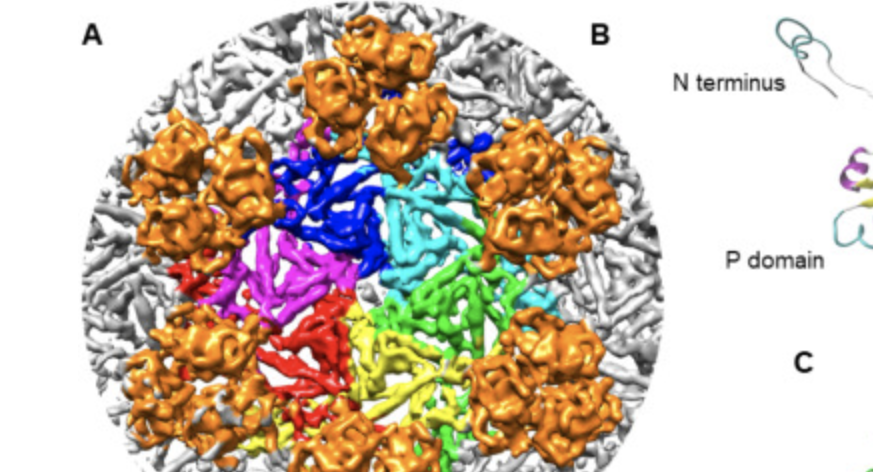
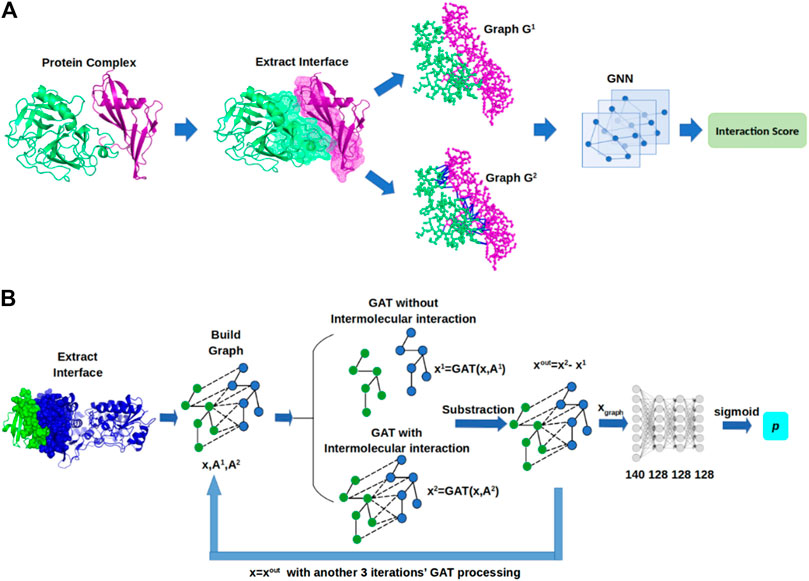
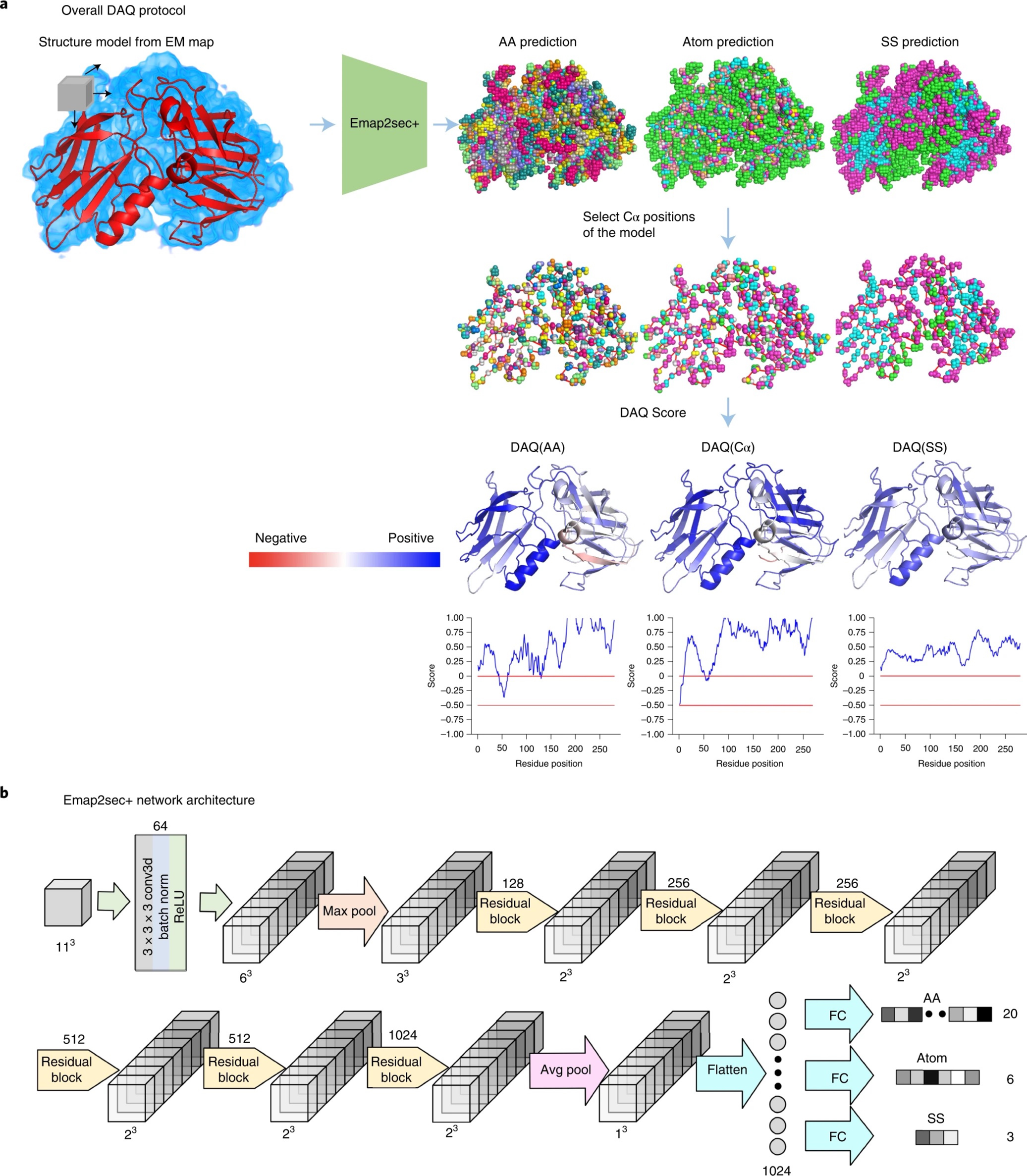

Specifically, we develop and apply novel computational methods for…
- Predicting protein structure from sequence
- Predicting protein function from sequence and structure
- Predicting protein-protein and protein-ligand interactions
- Predicting functional sites in sequences
- Genome-scale function and structure annotation
- Analyzing functional units in networks
More information can be found on our research projects page here
For information on lab openings, see here









